Deficiência de alfa-1 antitripsina
Sinônimos em um sentido mais amplo
- Síndrome de Laurell-Eriksson
- Deficiência de inibidor de protease alfa-1
Inglês: deficiência de alfa1-antitripsina
introdução
A deficiência de alfa-1-antitripsina significa, como o nome sugere, a falta da proteína alfa-1-antitripsina, que se forma nos pulmões e no fígado. Portanto, é um distúrbio metabólico.
Esta condição é herdada de forma autossômica recessiva. Ocorre com frequência de 1: 1000 a 1: 2500 na população.

causas
A causa da deficiência de alfa-1 antitripsina está em uma falha na herança.
A deficiência da proteína alfa-1-antitripsina é herdada de forma autossômica recessiva. Isso significa que a doença é herdada independentemente do sexo e só surge realmente se houver duas cópias defeituosas do gene. Ambos os pais devem estar doentes ou agir como portadores da informação genética. Apenas um único gene que carrega a informação incorreta não pode causar nenhum dano. O defeito está no cromossomo 14. Neste cromossomo está o gene que é usado em indivíduos saudáveis para a síntese (Manufatura) da alfa-1 antitripsina é responsável.
Alfa-1-antitripsina é uma proteína produzida pelo corpo (proteína), que é produzido principalmente nas células do fígado.
Ele tem a tarefa de inibir enzimas que dividem proteínas. Uma deficiência de alfa-1-antitripsina agora leva à atividade excessiva dessas enzimas de divisão de proteínas. Isso resulta na degradação do próprio tecido do corpo. Sua tarefa mais importante é inibir a enzima elastase leucocitária. Isso divide a elastase na parede dos alvéolos.
Sintomas e doenças
Uma vez que a produção de alfa-1-antitripsina ocorre principalmente nos pulmões e no fígado, danos e deficiências também ocorrem aqui. A degradação do próprio tecido do corpo também ocorre lá.
Existe uma grande variabilidade na forma. Em pessoas com lesões pulmonares graves, o envolvimento do fígado é surpreendentemente raro e vice-versa. A distribuição de idades também é bastante diferente. Enquanto alguns apresentam doenças pulmonares em estágio terminal da terceira à quinta década de vida, outros não apresentam nenhum dano aos pulmões aos 30 anos de idade.
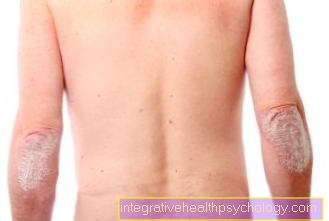
Sintomas na pele
Às vezes, os pacientes com deficiência de alfa-1 antitripsina apresentam inflamação na gordura subcutânea. Isso é demarcado e avermelhado. É chamado de paniculite. Existem outras causas para essa inflamação. O mecanismo exato de sua formação ainda não é conhecido. Essa inflamação localizada pode ser muito persistente e dolorosa.
Outro sintoma na pele é a cor azul (cianose). Isso é causado pela saturação insuficiente de oxigênio do sangue quando os pulmões estão envolvidos, como enfisema. Não só a pele fica com uma coloração azulada, mas também as membranas mucosas e a língua. A cianose ocorre em muitos quadros clínicos, por isso não é específica para a deficiência de alfa-1 antitripsina.
Sintomas e consequências nos pulmões
A proteína alfa-1-antitripsina não é encontrada apenas no fígado, mas também nos pulmões. Aqui também desempenha um papel importante na boa função pulmonar. Em caso de deficiência desta alfa-1 antitripsina, componentes importantes dos pulmões são degradados, resultando na destruição constante do tecido pulmonar.
Uma deficiência de alfa-1 antitripsina causa enfisema nos pulmões. O enfisema pulmonar é uma hiperinsuflação dos pulmões. Isso ocorre devido às alterações inflamatórias na estrutura pulmonar. As paredes dos alvéolos não são mais estáveis o suficiente e são destruídas pela degradação enzimática. Isso cria grandes cavidades nos pulmões, das quais o ar inalado não pode mais escapar. É por isso que se fala em hiperinsuflação dos pulmões.
A doença pulmonar obstrutiva crônica (DPOC) também se desenvolve no início da idade adulta. A troca gasosa nos pulmões é perturbada, resultando em falta de oxigênio no sangue. Uma tosse com expectoração é típica na DPOC.
A sensação de falta de ar também é típica no estágio avançado. Isso também pode ter consequências para o coração, de modo que ele também fica danificado. No caso de lesão pulmonar muito avançada e falha de outras medidas terapêuticas, um transplante de pulmão pode ser uma medida necessária.
Leia mais sobre: DPOC em estágio final
Sintomas e consequências no fígado
O fígado é o primeiro órgão a ser afetado pela deficiência de alfa-1 antitripsina. A proteína alfa-1-antitripsina está prejudicada. A forma da proteína é diferente da forma saudável. Isso leva ao fato de que ele se acumula nas células do fígado e não pode ser liberado adequadamente. Isso cria um defeito.
Em recém-nascidos que são homozigotos (ou seja, eles têm duas cópias de genes defeituosos) da doença, o fígado já está danificado na infância. Eles são diagnosticados com icterícia neonatal prolongada (icterícia = amarelecimento da pele e esclera (branco dos olhos)).
Se a doença aparecer apenas na idade adulta (aproximadamente 10-20%), é acompanhada por hepatite crônica (inflamação do fígado) e subsequente cirrose hepática.
Além disso, o risco de desenvolver câncer de fígado (carcinoma hepatocelular) é aumentado. A cirrose do fígado pode levar a muitas complicações para as pessoas afetadas. Em um estágio avançado, a expectativa de vida também é significativamente reduzida.
Leia mais sobre o assunto: Cirrose do fígado
diagnóstico
O diagnóstico de deficiência de alfa-1 antitripsina é baseado em uma amostra de sangue e exames laboratoriais. O sangue do paciente é examinado quanto aos seus componentes individuais (aqui, especialmente para a composição da proteína).
Há uma ausência quase completa de proteínas alfa-1. Enzimas hepáticas elevadas também podem ser encontradas no sangue. A ultrassonografia mostra um fígado aumentado (med.: Hepatomegalia).
Uma biópsia do fígado (amostras de tecido do fígado) também mostra depósitos característicos.
Quais são os valores do fígado com uma deficiência de alfa-1-antitripsina?
Uma vez que a enzima alfa-1-antitripsina não é formada corretamente no fígado, a enzima formada incorretamente é depositada nas células hepáticas e, portanto, as destrói.
Isso aumenta os marcadores de parênquima hepático, como GOT, GPT e glutamato desidrogenase (GLDH). A fosfatase alcalina também está freqüentemente aumentada. Com a cirrose hepática avançada, outros parâmetros também são afetados. Típico seria uma albumina diminuída, uma esterase de colesterol diminuída (CHE) e fatores de coagulação diminuídos, bem como um valor de amônia aumentado.
Leia mais sobre isso: Valores hepáticos elevados
Qual teste pode detectar a deficiência de alfa-1 antitripsina?
Existem dois testes que realmente comprovam essa condição. Essa é a eletroforese de soro e o teste genético.
Na eletroforese sérica, determina-se a concentração total de proteínas séricas do sangue e o fracionamento dessas proteínas. É um teste de diagnóstico laboratorial. Em geral, as concentrações de proteína são mostradas como uma linha com picos em um sistema de coordenadas. Existem 5 picos, o segundo pico desta curva mostra o conteúdo de alfa-1 globulinas, que inclui alfa-1 antitripsina. Se houver uma deficiência, esse pico é correspondentemente menor.
O teste genético é realizado, por exemplo, em um laboratório de genética humana. Para fazer isso, o DNA do paciente é examinado em busca de mutações no gene associado (ver herança).
Todos os outros testes, como um teste de função pulmonar, uma radiografia de tórax ou um ultrassom do fígado, podem explicar os sintomas da doença, mas não a causa.
Este artigo também pode interessar a você: O Teste Alfa-1 Antitripsina.
terapia
A deficiência de alfa-1-antitripsina agora pode ser facilmente sanada administrando-se a proteína por via intravenosa.
Além disso, no entanto, as doenças dos órgãos devem ser tratadas (especialmente a cirrose hepática) e qualquer dano já ocorrido deve ser reparado. Em casos extremos, entretanto, um transplante de fígado ou pulmão deve ser considerado.
A administração de alfa-1-antitripsina tem os seguintes efeitos colaterais:
- náusea
- Alergias
- febre
- Raro: choque anafilático (choque alérgico), que pode ser fatal
A terapia genética está em perspectiva no futuro.
Expectativa de vida
A deficiência de alfa-1 antitripsina se deve a várias mutações nos genes. É uma doença rara e hereditária que ocorre com uma frequência de cerca de 1: 2000 a 1: 5000.
As pessoas afetadas podem sofrer de uma forma leve ou grave da doença, que está associada a várias doenças secundárias e complicações.A expectativa de vida, especialmente daqueles pacientes que são afetados por uma forma grave, é reduzida em comparação com a população saudável. A expectativa de vida é estimada em cerca de 60 a 68 anos.
No entanto, isso se aplica apenas às pessoas afetadas que realizam terapia consistente e aderem a uma proibição estrita de fumar. O consumo de álcool também deve ser evitado, pois aumenta a probabilidade de ocorrência de doença hepática.
A expectativa de vida é fortemente dependente das doenças secundárias e da preservação da função orgânica dos pulmões e do fígado. No caso de falência de órgãos ou função severamente restrita, o último recurso geralmente é um transplante de órgão, que também está associado a uma expectativa de vida reduzida e ao risco de complicações adicionais.
profilaxia
Não há profilaxia real porque a doença é hereditária. As pessoas afetadas não devem fumar, pois isso torna mais difícil e coloca ainda mais pressão sobre os pulmões. O álcool também deve ser evitado devido à pressão sobre o fígado.
A deficiência de alfa-1 antitripsina é hereditária?
A deficiência de alfa-1 antitripsina é hereditária. A sequência do gene correspondente desta enzima está no 14º cromossomo.
Se a sequência do gene contém uma mutação, a sequência não pode mais ser lida corretamente e a enzima é formada incorretamente. A gravidade da doença é, portanto, variável. A mutação é herdada, ou seja, transmitida da mãe ou do pai. Um paciente está totalmente desenvolvido quando um defeito é herdado tanto do lado paterno quanto do lado materno. A extensão em que o paciente é afetado depende da genética, mas também de fatores externos, como o tabagismo.
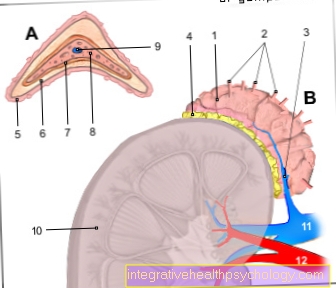
Anatomia e localização dos pulmões
- Pulmão direito
- Traquéia (Traquéia)
- Bifurcação traqueal (carina)
- Pulmão esquerdo
Resumo
A deficiência de alfa-1 antitripsina é uma doença metabólica hereditária, que resulta principalmente em alterações no tecido pulmonar. A frequência da doença é 1: 2000. Devido à falta dessa enzima, não há efeito inibidor sobre as enzimas de divisão de proteínas.
Devido a essa deficiência, seu próprio tecido pulmonar é decomposto ou digerido.
Ocorre enfisema pulmonar (incluindo tosse e falta de ar) e, com envolvimento adicional do fígado (10-20%), hepatite (icterícia). O diagnóstico é feito por meio de análise de sangue. A terapia é através da terapia de substituição, ou seja, a alfa-1-antitripsina é administrada artificialmente. A proteína em falta é administrada por via intravenosa (pela veia). Não há profilaxia.